

Regulatory submissions represent the cornerstone of pharmaceutical product development and commercialization, serving as the formal channel of communication between pharmaceutical companies and regulatory authorities.
According to recent industry analyses, the global pharmaceutical regulatory affairs market is valued at USD 9.47 billion in 2024 and is projected to grow at a CAGR of 7.17% from 2025 to 2030, highlighting the increasing importance and complexity of the regulatory landscape.
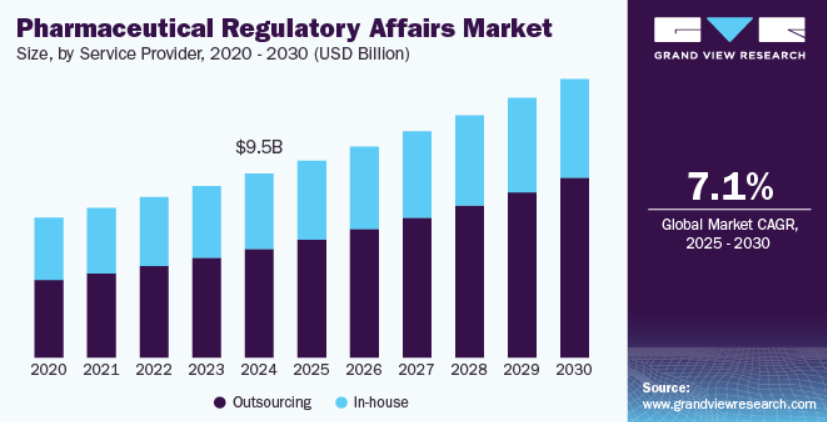
Source- Grandview Research
For pharmaceutical and biotech organizations, navigating the intricate web of regulatory submissions is not merely a compliance requirement but a strategic imperative that directly impacts time-to-market, product lifecycle management, and ultimately, patient access to life-saving therapies.
Understanding Pharmaceutical Regulatory Submissions
What Are Regulatory Submissions?
Regulatory submissions are comprehensive packages of scientific information and data submitted to regulatory agencies to demonstrate that a healthcare product is safe, effective, and manufactured to high-quality standards. These submissions occur throughout a product’s lifecycle, from initial clinical testing through post-marketing surveillance.
The Strategic Importance of Regulatory Filings
Beyond mere compliance, effective regulatory submission management offers strategic advantages:
- Market access acceleration: Well-prepared submissions can reduce review cycles and expedite approvals
- Competitive advantage: Organizations skilled in navigating regulatory requirements bring products to market faster
- Risk mitigation: Comprehensive submissions address potential safety and efficacy concerns preemptively
- Global market penetration: Strategic submission planning facilitates multi-regional approvals
Types of Regulatory Submissions in the US and EU
Pharmaceutical companies must navigate different submission types throughout a product’s lifecycle. Understanding these submissions is essential for efficient regulatory planning and compliance.
Pre-Marketing Submissions
- Clinical Trial Applications
- US: IND (Investigational New Drug) to FDA
- EU: CTA (Clinical Trial Application) to national agencies
- Includes preclinical data, manufacturing details, and clinical protocols.
- CMC Amendments
- Updates on manufacturing changes, analytical methods, stability data, and specifications.
- Ensures regulators stay informed on product quality.
- Marketing Applications
- US: NDA (New Drug Application), BLA (Biologics License Application)
- EU: MAA via centralized, decentralized, mutual recognition, or national procedures
- Includes clinical data, manufacturing, labeling, and risk plans.
Post-Approval Submissions
- Application Modifications
- US: PAS, CBE, annual reports
- EU: Type IA/IB/II variations depending on impact level
- Agency Inquiries
- US: Complete Response Letters
- EU: List of Questions, info requests
- Timely responses are key to avoiding delays.
- Post-Approval Commitments
- Includes post-marketing studies, PSURs/PBRERs (EU), and REMS (US)
- Supports ongoing safety and compliance.
Key Components of Successful Regulatory Submissions
Documentation Quality and Structure
Successful submissions depend on meticulously prepared documentation that follows agency-specific formats:
- Common Technical Document (CTD) format organization:
- Module 1: Regional administrative information
- Module 2: Summaries (quality, nonclinical, clinical)
- Module 3: Quality (CMC) information
- Module 4: Nonclinical study reports
- Module 5: Clinical study reports
- Electronic submission requirements:
- eCTD (electronic Common Technical Document) specifications
- Proper document granularity and lifecycle management
- Adherence to technical validation criteria
Data Integrity and Compliance
Regulatory submissions must demonstrate adherence to:
- Good Manufacturing Practices (GMP): Quality systems ensuring consistent production and control
- Good Laboratory Practices (GLP): Standards for nonclinical safety studies
- Good Clinical Practices (GCP): Ethical and scientific quality standards for clinical research
Data integrity principles must be maintained across all submission content:
- Attributable
- Legible
- Contemporaneous
- Original
- Accurate
Regulatory Strategy Development
A well-defined regulatory strategy enhances submission success:
- Target product profile alignment with submission content
- Health authority interactions (pre-IND/scientific advice meetings)
- Submission pathway selection (standard, accelerated, conditional, etc.)
- Global harmonization considerations across multiple markets
FDA vs. EMA: Key Differences in Regulatory Approach
While there is increasing harmonization between major regulatory agencies, important differences remain that impact submission strategy:
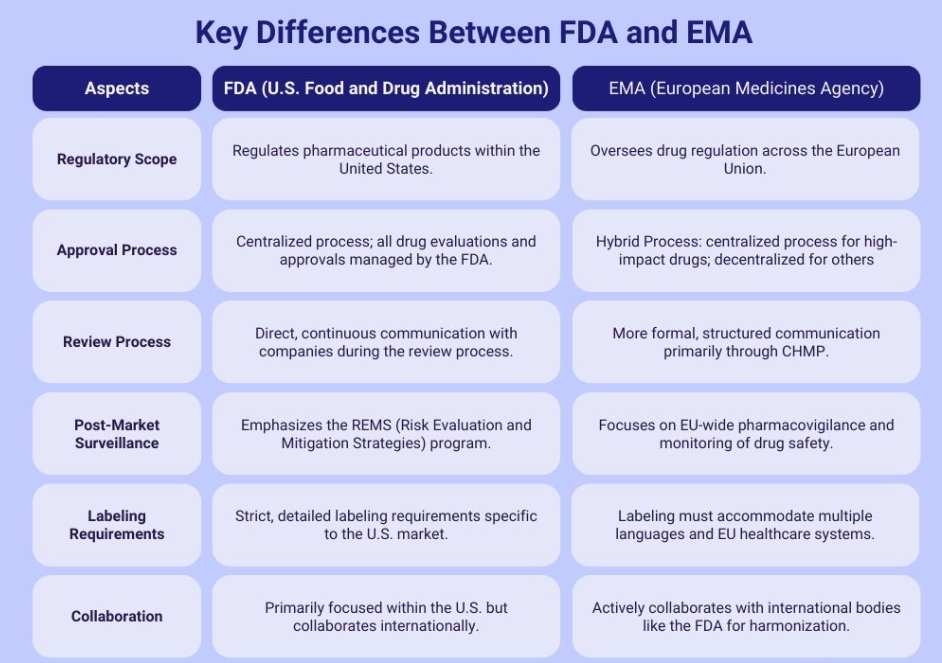
Understanding these differences allows for strategic submission planning that can accommodate both markets efficiently.
Source- Adragos Pharma
Common Challenges and Best Practices in Regulatory Submissions
Common Submission Pitfalls
- Inconsistent data presentation across sections
- Incomplete responses to previous regulatory questions
- Inadequate justification for manufacturing or clinical decisions
- Poor document quality (formatting, hyperlinks, bookmarks)
- Insufficient risk assessment for identified issues
Best Practices for Submission Success
- Plan Strategically: Build realistic timelines, perform early gap analyses, and ensure clear cross-functional communication.
- Maintain Documentation Rigor: Conduct thorough reviews, track requirements, and ensure data consistency across modules.
- Leverage Regulatory Insights: Stay current with guidance updates, study precedent approvals, and apply relevant advisory feedback.
- Use Smart Tools: Implement document management and publishing systems that support eCTD compliance and team collaboration.
Future Trends in Pharmaceutical Regulatory Submissions
Digital Transformation
The regulatory space is rapidly evolving with the integration of digital technologies. Artificial intelligence is being used to enhance submission consistency, while natural language processing accelerates automated document review. Cloud-based platforms are enabling real-time collaboration among global teams, and regulatory agencies are beginning to adopt systems that allow seamless, real-time data exchange with sponsors.
Global Harmonization Initiatives
Regulatory bodies worldwide are working together to streamline and align their processes. Initiatives like the International Council for Harmonisation (ICH) and the International Pharmaceutical Regulators Programme (IPRP) are setting unified technical standards. Programs such as Project Orbis are facilitating coordinated oncology product reviews, and reliance pathways are gaining traction as agencies increasingly leverage each other’s assessments.
Evolving Submission Requirements
Regulatory expectations are shifting to accommodate new scientific and clinical models. There is growing emphasis on integrating real-world evidence, supporting patient-focused drug development, and meeting specialized requirements for advanced therapies such as cell and gene treatments. Additionally, decentralized clinical trial models are influencing submission formats and data requirements.
Key Takeaways
- Regulatory submissions are strategic assets that directly impact market access and competitive positioning
- Understanding submission types and requirements across major markets enables efficient regulatory planning
- Successful submissions depend on quality documentation, data integrity, and cross-functional collaboration
- Differences between FDA and EMA approaches necessitate tailored submission strategies
- External regulatory support can provide specialized expertise for complex submission challenges
- Digital transformation and harmonization initiatives are reshaping the future of regulatory submissions
FAQ: Pharmaceutical Regulatory Submissions
What is the difference between an IND and an NDA?
An Investigational New Drug (IND) application requests permission to begin human clinical trials for an experimental drug, while a New Drug Application (NDA) seeks marketing approval based on completed clinical trials demonstrating safety and efficacy.
How long does the regulatory review process typically take?
Review timelines vary by submission type and region. In the US, standard NDA/BLA reviews typically take 10-12 months, while priority reviews may be completed in 6 months. In the EU, the standard MAA review takes approximately 210 active days, though clock stops during information requests can extend the total calendar time.
What are the key components of an eCTD submission?
An eCTD (electronic Common Technical Document) submission includes five modules: regional administrative information (Module 1), summaries (Module 2), quality data (Module 3), nonclinical study reports (Module 4), and clinical study reports (Module 5), all organized according to a standardized electronic structure with proper metadata.
How do orphan drug designations affect regulatory submissions?
Orphan drug designations for rare diseases offer several regulatory advantages, including reduced fees, protocol assistance, market exclusivity, and potentially streamlined review processes. However, submission content requirements for demonstrating safety and efficacy remain rigorous.
What are the most common reasons for regulatory submission rejections?
Common reasons for submission rejections include insufficient efficacy data, unexpected safety concerns, manufacturing quality issues, inconsistencies in data presentation, inadequate risk management plans, and failure to address previous regulatory questions comprehensively.
How are post-approval changes managed from a regulatory perspective?
Post-approval changes are managed through variation/supplement submissions whose categorization (minor vs. major) depends on the change’s potential impact on product quality, safety, and efficacy. Changes may require prior approval or simple notification depending on their classification and regional requirements.
What role does pharmacovigilance play in regulatory submissions?
Pharmacovigilance information features prominently in initial marketing applications (risk management plans) and post-approval submissions (periodic safety reports, signal evaluations). Robust pharmacovigilance systems and data are essential for maintaining regulatory compliance throughout a product’s lifecycle.
Recent Regulatory CDMO News-
AGC Biologics Seattle Completes FDA Inspections and Secures Global Regulatory Approvals (Sept 2025)
Symeres Acquires DGr Pharma to Enhance Regulatory Expertise (Sept 2025)
Wheeler Bio Partners with Pharmefex to Strengthen IND Regulatory Support (Sept 2025)
Facet Life Sciences Named Regulatory Partner for Nucleus Radiopharmaceuticals (May 2025)
DCN Dx Expands Services with New Regulatory Affairs Offering (March 2025)
Boehringer Ingelheim Expands Contract Manufacturing in China with Regulatory Reform (March 2025)
Download our CDMO News Tracker to stay ahead of every shift in the CDMO landscape.






