“If I could have a magic wand to increase this awareness, I would — so that everybody understands the necessity to go through the details right at the beginning of commercialization.”
Alexander Huber is Global Head of Launch Excellence and Digitalization for Specialized Modalities at Lonza, where he leads the team responsible for guiding late-stage cell and gene therapy programs through commercialization across Lonza’s global network of manufacturing sites. He spoke with PharmaSource at the Advanced Therapies Congress in London in March 2026.
In this interview, Alexander draws on his firsthand experience commercializing some of the earliest approved cell therapies to identify where programs most commonly run into trouble — and what needs to happen differently, earlier.
Cell and gene therapy has now produced a small but meaningful number of approved commercial products. The field is no longer purely experimental. But the path from a successful clinical readout to reliable commercial supply remains one of the most technically and operationally demanding transitions in pharmaceutical development — and one that is still routinely underestimated by the teams navigating it.
Alexander has spent more than a decade working on that transition, including hands-on experience with Kymriah, one of the first approved CAR-T therapies, across manufacturing sites in Japan, China, and Europe. His current role at Lonza is built around the systematic challenges that recur across programs regardless of the specific therapy or indication: the gaps that appear when clinical-stage assumptions meet the realities of commercial-scale GMP manufacturing.
His core message was that the upfront work required to commercialize a cell or gene therapy program is consistently underestimated, and the consequences of that underestimation — roadblocks discovered late, timelines lost, batches at risk — are avoidable if the right framework is in place early enough.
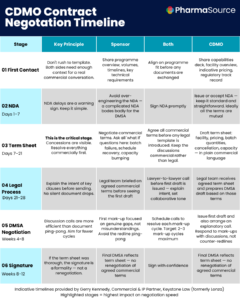
Three Areas That Consistently Catch Programs Off Guard
When Alexander talks about what goes wrong during CGT commercialization, he returns to three areas with notable consistency: scale-up demonstration, analytics robustness, and raw material control. Each of them tends to be manageable at clinical volumes. Each of them becomes a different problem entirely at commercial scale.
Scale-up is the most visible challenge, particularly for autologous therapies where one patient equals one batch and the manufacturing rhythm has to increase without any of the traditional levers — larger vessels, pooled materials, extended campaigns — available to conventional pharma. Regulatory authorities, and the FDA in particular, require manufacturers to demonstrate that they can actually deliver at commercial volumes. That demonstration takes time, resources, and advance planning that many programs do not allocate.
“Typically, you are in clinical manufacturing, which is low volumes — one or two batches per month, maybe. And then you need to go to scale-up. Increasing this rhythm, and having to demonstrate the capacity to authorities — this is usually underestimated. It is a lot of work.”
Analytics is the second pressure point. At clinical scale, an analytical method that is not fully robust is manageable — the volume of releases is low, and problems can be absorbed. At a commercial scale, the same method has to release hundreds of batches reliably. Any weakness in the analytical package that was tolerable during clinical development becomes a patient safety and supply continuity issue at commercial volumes.
Raw materials is where Alexander’s experience leads him to be most emphatic. Cell and gene therapy programs can involve hundreds of individual raw materials, many of them highly specialized — viral vectors, antibody-coated beads, plasmid DNA — with limited supplier options and long lead times. Managing that supply chain at clinical scale is challenging. Managing it at commercial scale, with the supply continuity requirements that commercial GMP demands, is an order of magnitude more complex.
“Raw materials are always underestimated. In that area, you typically have hundreds of raw materials. A lot of them are critical, and everything needs to be under control. Not as in standard pharma where you have maybe ten raw materials. You have to have your supply chain under control.”
Lonza’s response to this is to move toward dual sourcing for critical materials wherever possible, and to qualify more robust primary sources earlier than programs typically plan for. The instinct to defer those conversations on the grounds that clinical supply is running and commercial is still some way off is, in Alexander’s view, one of the more expensive mistakes a program can make.
Why GMP Is a Different Animal Than Science
One of the more consistent patterns Alexander observes is the gap between scientific confidence and GMP readiness in leadership teams approaching commercialization for the first time. Scientists who have driven a therapy through clinical development have done something extremely difficult. The assumption that regulatory bodies will recognize the scientific merit of what they have achieved — and that the path to approval will follow naturally — is understandable, but it does not reflect how commercialization actually works.
“What we typically see is that scientists are fascinated by science, and they think: this is a great therapy, the regulators will see the benefit automatically. But no — this is almost never the case. You need to prove everything, and it goes into so much detail. GMP is such a different animal from science. It is regulated, it’s boring, cumbersome — you need to describe everything, everything needs to be documented. And this is usually really underestimated by the scientific side.”
The discipline that commercial GMP manufacturing requires — documentation at every step, validation of every process, audit trails for every deviation — is not a bureaucratic overhead that exists separately from the science. It is the mechanism by which regulators and, ultimately, patients can trust that the product is what it is claimed to be, batch after batch. For programs that have operated in a clinical mindset throughout development, building that discipline into the organization before commercialization begins is a significant undertaking.
Alexander’s advice to biotech CEOs approaching this transition is to engage with GMP experts early as a strategic input to how the commercialization plan is structured. The organizations that treat GMP readiness as a later-stage problem consistently discover it later, at greater cost.
Automation Is Changing the Manufacturing Model
A significant part of Lonza’s current focus in cell and gene therapy is moving programs away from manual processing toward automated manufacturing. Alexander describes this shift as one of the most consequential changes currently underway in the field — both for cost and for the practicality of delivering autologous therapies at commercial volumes.
“A lot of the cell and gene processes are still quite manual. So everybody is investing a lot in optimization. If you can move away from high labor intensity to more automated processes, it brings down the costs.”
The model he describes moving toward is what he calls a ballroom concept: multiple automated processing units operating in parallel within a single room, managed by a single operator rather than multiple operators per individual step. For autologous therapies, where one patient always equals one batch and that constraint will not change, this approach offers a meaningful improvement in throughput and cost without compromising the chain-of-identity requirements that patient-specific manufacturing demands.
“It’s always one batch in one system. This will not change. But you can do a ballroom with multiple units in one room.”
Underpinning that model are two enabling requirements Alexander is explicit about: automation systems and IT infrastructure, specifically electronic batch record systems, that can ensure chain-of-identity is maintained even when multiple patient batches are being processed in parallel in the same space. At commercial volumes, spreadsheet-based approaches are no longer adequate. Lonza now offers customers API-connected access to their manufacturing process data directly, replacing periodic manual reporting with continuous data access.
Actionable Takeaways
- Start your commercialization mapping before your pivotal data readout. The work required to demonstrate commercial-scale capacity, validate analytics at volume, and secure critical raw materials takes longer than most programs plan for. Beginning that mapping while clinical data is still maturing is not premature.
- Audit your analytical methods against commercial release volumes now. A method that works at clinical scale may not be robust enough to release hundreds of batches reliably. Identifying and addressing those gaps before you need the method for commercial release is substantially less disruptive than discovering them after.
- Dual-source your critical raw materials before you need commercial volumes. Qualifying a second supplier for viral vectors, plasmids, or antibody-coated beads takes time. Start that qualification process earlier than feels necessary.
- Treat GMP readiness as a strategic input, not a compliance function. Bring GMP expertise into commercialization planning conversations early. The organizations that treat it as a later-stage problem consistently encounter it as a later-stage crisis.
- Evaluate your chain-of-identity and batch record systems against commercial requirements. Spreadsheet-based approaches that work at clinical volumes will not scale to commercial autologous manufacturing. Plan your digital infrastructure upgrade as part of commercialization planning.
Outlook
The cell and gene therapy field has demonstrated that it can produce approved, commercially available therapies. What has not yet been fully resolved is the systematic underestimation of what the clinical-to-commercial transition actually requires. Alexander’s experience — across Kymriah’s launch in multiple markets and now across Lonza’s broader portfolio of late-stage programs — is that the gap is not primarily technical. The science, in most cases, is sound. The gap is in the planning discipline that commercialization demands and that clinical-stage organizations are rarely structured to provide.
The programs that will reach commercial supply reliably, on time, and at a cost that makes the therapy financially viable are the ones that start that planning early, map the full journey before they begin it, and treat GMP manufacturing as an equal partner to the science that made the therapy possible in the first place.