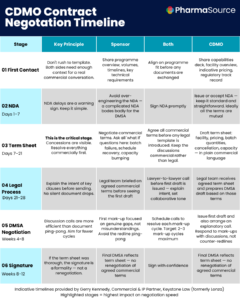
“I wish academics would start research projects with the end of the translational journey in mind — thinking about how every choice you make at the beginning affects the entire life cycle of getting a therapy to a patient.”
Toby Gamlen is Head of Manufacturing at the Gene Therapy Innovation and Manufacturing Centre (GTIMC) at the University of Sheffield, an academically rooted, client-driven facility producing AAV vectors for researchers translating novel therapeutics toward first-in-human trials. He spoke with PharmaSource at the Advanced Therapies Congress in London in March 2026.
In this interview, Toby draws on his experience guiding academic gene therapy programs through the earliest stages of translational development to identify where the gap between research and GMP manufacturing most commonly opens up — and what researchers and early-stage biotech leaders can do to close it before it becomes expensive.
The Gene Therapy Innovation and Manufacturing Centre sits at an unusual point in the cell and gene therapy ecosystem. It is not a commercial CDMO in the conventional sense. It is a non-profit, academic facility, part of the Innovation Hubs for Gene Therapy project, funded by the Medical Research Council and LifeArc, with support from the Biotechnology and Biological Sciences Research Council, but it operates in a client-driven, service-oriented model, manufacturing AAV vectors at cost for academic groups working toward first-in-human trials. The programs it works with are earlier than those typically served by larger commercial manufacturerswhich puts Toby’s team at the point in the pipeline where the gap between academic research and GMP manufacturing is most visible.
What he observes, consistently, is that the decisions researchers make years before they engage with a manufacturing partner — choices about data collection, vector design, proof-of-concept methodology — frequently create problems that are expensive and time-consuming to resolve when the program eventually needs to scale. The gap is not primarily technical. It is a gap in awareness: researchers who have spent a decade or more developing a scientific understanding of a disease and a potential therapeutic approach often have not been exposed to what GMP manufacturing actually requires, and the consequences of that gap compound over time.
The Decisions Made Ten Years Ago That Cause Problems Today
The translational pipeline for academic gene therapy programs is often long, frequently spanning a decade or more from foundational research to first-in-human trials. Engagement with manufacturing typically begins during late preclinical development, though the underlying science may have been in development for many years prior. The choices made at the beginning of that journey — about how data is collected and documented, about the scale at which proof-of-concept work is conducted, about the vector and delivery system selected — all have downstream consequences that are not visible at the time but become significant when manufacturing scale-up begins.
“For a lot of those academics, it might be a lifetime’s worth of work getting to the point where they’re starting to engage with this,” Toby explains. “The choices they made ten, fifteen years ago in their research life cycle can undermine them later on. It’s just exposure. If academics aren’t exposed to that world early on, they might not make the right choices.”
One of the most common practical problems Toby’s team encounters is researchers arriving with a vector that was fit for purpose at proof-of-concept scale, but is not suitable for the larger scale toxicology work or manufacturing runs required for clinical translation. The initial vector may have been purchased from an external supplier, produced under research conditions, or designed with parameters that made sense for laboratory work but create significant complications when the program needs to demonstrate manufacturing feasibility at GMP scale.
“They might have purchased a vector from a supplier, and it’s not really fit for purpose for a larger-scale toxicology study. So suddenly they’re having to go through the feasibility elements of taking their initial proof-of-concept vector and going: right, now you need to produce this at a scale sufficient for tox work and large scale production. And those systems just don’t work together — you suddenly have to start revisiting earlier decisions, which takes more time.”
Time and cost are the direct consequences, and in Toby’s experience, the two are inseparable. Feasibility studies at small scale, analytics work, the iterations required to bring a research-grade process up to GMP standards — all of these take longer and cost more than academic programs typically plan for, precisely because they were not anticipated at the outset.
What Researchers Should Do Differently From the Start
When Toby is asked what advice he would give to an academic group at the earliest stages of translational development, his answer is structured around four practical steps — each of which addresses a specific category of problem he has seen derail programs later.
The first is data integrity. Every experiment, from the earliest stages of research, should be conducted and documented in a way that will be defensible in a GMP context later. Engaging with consultancy early to understand what data control standards will eventually be required is considerably less disruptive than attempting to reconstruct or revalidate data that was collected without those standards in mind.
The second is cost modeling. The jump in expenditure when a program moves from research-scale to GMP production is significant and consistently underestimated. Understanding what clinical-scale manufacturing will cost — for the vector, for analytics, for the full process — and building that into financial planning early gives programs a more realistic picture of what reaching first-in-human actually requires.
The third is regulatory engagement. Toby’s advice is to begin talking to regulators as soon as there is a physical product to discuss. The regulatory requirements for a first-in-human gene therapy trial are detailed, and the timelines are long. Early engagement reduces the risk of discovering a regulatory gap at a point where it can delay the program significantly.
The fourth is foundational discipline in the research process itself — an area where Toby’s experience has produced some surprises. “The basics — like making the switch away from ampicillin resistance genes in plasmids, making sure that what you’re doing early on in the translational pathway won’t create problems for a product intended, ultimately, for a patient.”
The common thread across all four is the same: start thinking about the requirements of the end state — GMP manufacturing, regulatory submission, clinical trial — from the beginning of the research process, not when the program arrives at a manufacturing facility.
AAV Manufacturing Capacity
Beyond the immediate challenges facing individual programs, Toby has a broader view on the structural state of AAV manufacturing capacity that is relevant to anyone planning a gene therapy development program today.
His assessment is that there is meaningful manufacturing capacity developing in the sector, but that it is fragmented and disconnected in ways that limit its collective value. Multiple facilities — academic and commercial — are building capability independently, without the kind of data sharing, best practice exchange, or coordinated development that would allow the field to progress faster.
“There’s a lot of disconnected manufacturing capacity. I’d be really interested in seeing people bring together consortia of manufacturers who aren’t necessarily within the same organization — forming groups where best practice can be shared, and you can start to see greater impact from small facilities working together.” The Innovation Hubs for Gene Therapy is one example of this nationally, and then at the European level, the GeneT project from the University of Coimbra, which we are contributing to, is another great example.”
AI-assisted process development and quality assurance are areas where the field could make meaningful progress — but only if the underlying data sets are large enough to train reliable models on. Individual facilities, particularly smaller ones, do not generate sufficient data independently to build those models. Shared data across a network of manufacturers could accelerate process development, quality analytics, and regulatory understanding in ways that siloed facilities cannot.
“We don’t have the data sets as individual organizations unless you’re one of the massive organizations. The more that we work together, the faster we generate the data sets that we can train AI on to really accelerate development.”
This is not purely idealistic. There are working models for it. Toby points
to the respiratory gene therapy field, where charitable funders have incentivised collaboration between previously competing research groups. The result was a program that reached first-in-human trials. Applying a similar funding-driven incentive model to manufacturing consortia, at least in the earlier, pre-commercial stages of development, is something Toby believes could meaningfully accelerate the field.
Big Pharma Pullback
The reduction in large pharma investment in gene therapy over recent years is a topic that generates significant anxiety in parts of the field. Toby’s reading of it is more measured. He frames it as a stabilization — a correction after a period of unrealistic expectations about the speed at which blockbuster gene therapies would reach commercial scale — rather than a structural retreat from the modality.
“I think it’s an opportunity because there is a realism coming into the sector. It’s harder to source funding, but it’s much more realistic about which products and systems will fly in the future. And it creates niches.”
The reimbursement model for gene therapies, particularly in European markets, has proven more challenging than early projections assumed. The result is that the programs most likely to succeed commercially in the near term are not the large-scale blockbuster indications that attracted the most investment, but smaller, more focused programs targeting ultra-rare disorders — where the patient population is small, the unmet need is high, and the manufacturing volumes are correspondingly manageable.
For early-stage programs and the academic facilities supporting them, that is not necessarily unwelcome news. Smaller patient populations mean smaller manufacturing requirements, which means more of the capacity being developed at facilities like the GTIMC is directly relevant to the programs most likely to reach patients in the current environment.
Actionable Takeaways
- Engage with manufacturing and regulatory consultancy at the earliest stages of your research program. The decisions you make about data collection, documentation, and vector design at proof-of-concept stage will either facilitate or complicate your path to GMP manufacturing. Getting that guidance early costs less than correcting problems later.
- Build a realistic cost model for GMP production before you need it. The jump from research-scale to GMP manufacturing cost is significant and consistent. Programs that have not modeled it early routinely discover funding gaps at the worst possible moment.
- Start regulatory conversations as soon as you have a physical product to discuss. Regulatory timelines for first-in-human gene therapy trials are long. Early engagement with regulators is protective, not premature.
- Check that your proof-of-concept vector is scalable before you build your program around it. A vector that was fit for laboratory-scale work may not be suitable for the volumes required for toxicology studies or GMP manufacturing. Verifying scalability early avoids a significant and time-consuming course correction later.
- Seek out manufacturing networks and consortia rather than navigating the translational path in isolation. The field is developing shared knowledge and best practices faster than any individual program can access independently. Conference engagement, CDMO conversations, and collaborative funding models all offer access to that knowledge.
What Toby’s perspective brings into focus is a part of the gene therapy development pipeline that is often invisible in conversations about manufacturing capacity, CDMO selection, and clinical scale-up: the earliest translational stage, where the choices that will determine whether a program ever reaches manufacturing are actually made.
The academic researchers working at that stage are, in many cases, doing important science. What they frequently lack is exposure to the manufacturing, regulatory, and commercial realities that will govern whether that science can reach a patient. Closing that gap earlier, through better education, earlier consultancy engagement, and more collaborative working across the sector, is not a peripheral concern for the gene therapy field. It is where a significant proportion of future programs will be won or lost.